发布时间:2023-01-06
来源:精准医学临床实验室
一、简介
听力障碍或耳聋是最广泛的遗传性感觉障碍之一,每1000个新生儿中有1-2个受到影响。大约三分之一的遗传性听力损失(HHL)病例是伴随着其他医疗异常而被诊断出来的,因此被归类为综合征性耳聋。约三分之二的病例是非综合征性听力损失(NSHL)型,其中80%是先天性的,而20%是语言后发病。在大多数情况下,听力损失的原因是感音神经性听力损失(内耳或前庭耳蜗神经缺陷)。
在胚胎发育过程中,耳朵结构的形成涉及到区域(或轴向)极性的形成。受精第4周后,在发育中的胚胎内陷形成内耳,随后形成耳泡,接着形成膜迷路,最后发展为内耳结构。在耳朵元件的轴向定位过程中,各种蛋白质、转录因子和形态因子介导了上皮细胞和间充质细胞之间的相互作用。参与Wnt、Notch、FGF、BMP和Shh信号通路的基因负责调控这些复杂的相互作用。无用的非必要组织和受损毛细胞会通过凋亡途径去除。
在过去的几十年里,大量涉及上述途径的基因的致病性变异都与非综合征性听力损失NSHL相关。本次系统综述的目的是:
i) 汇编迄今为止已报道的导致NSHL的单基因突变;
ii) 阐明所涉及基因的突变频率;
iii) 报告迄今为止所研究的种族/人群。
二、统计结果
综述统计筛选了271篇论文中的病例,包含来自世界各地的2846名患者和来自中国的12个核心家庭。其中大部分的患者属于东亚地区。男性960例,女性906例,其余病例为性别未知。除12例外,所有受试者均有双耳听力损失。数据分析显示,1722例语言前病例和1010例语言后病例,说明语前发病情况可能更为常见,且许多病例的发病年龄仍未确定。这些数据进一步表明,常染色体隐性NSHL通常是语前和先天性的,而常染色体显性NSHL多有语后听力障碍的家族史。此外,许多语言后发病的听力损失病例都是由于线粒体遗传变异导致的,属于母系遗传。
(1) NSHL的单基因致病变异
经统计共有98个基因含有导致NSHL的单基因致病性突变(表1)。其中GJB2是最常见的基因,与849例患者相关,最常见的GJB2基因变异类型为35delG,与132例患者相关。此外,在271项已报道的研究中,有156项研究提到首先筛选了GJB2,74项研究报道了新的候选基因和功能研究。然而,根据138项研究,在2846名患者中有1324个个体(548名男性,532名女性,其余性别未指明)中报道了GJB2以外的致病基因。这138项研究没有报告功能研究,而是依赖于先前已知的研究。通过9项深入的连锁分析,确定了潜在的候选位点。这些研究共包括100名患者,其中41名男性和59名女性。
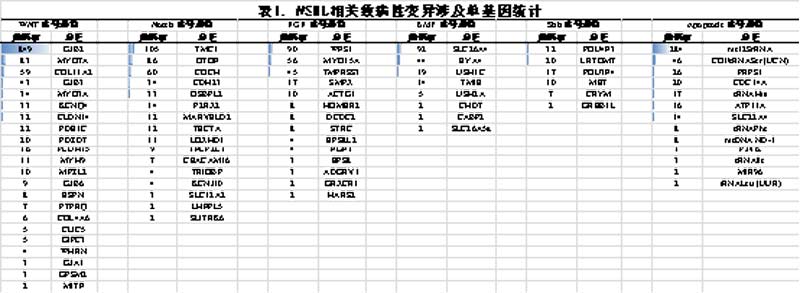
(2) NSHL相关的单基因频率
研究已报道在849例患者中发现GJB2基因的致病变异,使其成为NSHL最常见的相关基因。此外,在105个先证者中报道了TMC1基因的致病变异,在92个先证者中报道了SLC26A4基因的致病变异,在90个先证者中报道了WFS1基因的致病变异。在线粒体基因组中,研究发现142例患者的mt12SrRNA基因存在致病性遗传变异。研究中报道3例或者少于3例的致病变异相关基因有CHD7、ILDR1、GREB1L、CABP2、GSDME、SLC26A5a、CIB2、PEX6、SLC12A2、EPS8、GRXCR1、HARS2、TBC1D24、MIR96、tRNALeu、SYNE4和GPSM2。其他仅报道过1例致病变异相关基因有PAX3、DSPP、MYH14、GRHL2和ESRRB;大多数这种罕见的变异仅在一个家庭中报道过。在2例MYO1A突变的患者中,一名来自韩国的女性患者没有其他相关突变,而另一名来自伊朗的男性患者存在PTPRQ基因变异,具有复合杂合子特征。
三、NSHL相关的基因功能
(1)在人类胚胎耳朵发育中的作用
研究NSHL相关基因的致病性遗传变异信息有助于深入了解胚胎的早期发育途径以及遗传变异对于听觉器官形成的影响。以往认为耳聋相关基因可能在多种分子通路中发挥作用,但这些基因突变导致的非综合征性耳聋除了听力损失外没有观察到其他临床特征,表明这些基因最重要的作用仅在耳朵发育和听力。因此,深入了解这些基因突变所影响的分子途径将为将来治疗这种疾病提供更多靶点。
耳朵的胚胎发育涉及一系列复杂的事件,包括所有三种类型的胚层(外胚层、中胚层和内胚层),以及诱导信号的及时作用,从而最终产生一个功能性的听觉器官。感音神经性听力损失是由于听觉神经或内耳的缺陷。在内耳早期胚胎发育过程中,某些蛋白质使多种信号通路相互作用和相互连接。这些信号通路包括Wnt、Notch、Shh、FGF、BMP和凋亡通路(图2)。
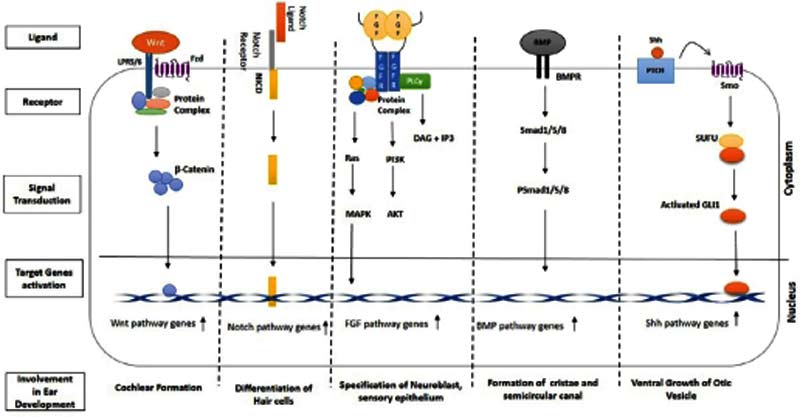
(2) Wnt信号通路
本文共统计到至少在2例患者中出现的22个Wnt信号通路基因的致病性变异。这些基因大多具有维持支持细胞之间的间隙连接、离子细胞和运动以及静纤毛的调节特性。Wnt网络由典型的、非典型的(Planer Cell Polarity,PCP通路)和Wnt/钙信号通路组成,其中典型的信号通路参与了感觉上皮细胞的细胞命运调控。这一通路在耳部发育的早期阶段仍然被激活,并在背侧命运(前庭系统)和腹侧命运(耳蜗)之间划分了耳部前体。另一方面,PCP通路参与调节细胞-细胞重排或细胞骨架重组(在静纤毛束形成过程中),并且在毛细胞的发育和静纤毛的单向组装中发挥关键作用。
(3) Notch信号通路
Notch通路在内耳发育中是从外胚层的形成开始起作用,外胚层是内耳形成的早期阶段。该通路调节毛细胞和内耳支持细胞的分化,并介导感觉区域的模式形成。此过程通过Notch配体(存在于分化的毛细胞中)和Notch受体的相互作用来实现。目前研究共发现16个Notch信号通路相关基因的致病性变异与NSHL有关。
(4) FGF信号通路
共有14个FGF(Fibroblast Growth Factor)信号通路基因的变异与NSHL有关。FGF信号通路增强了wnt介导的细胞增殖,反之亦然。但与Notch信号通路一样,FGF信号通路也通过下调Atoh1(负责支持细胞向毛细胞分化)来抑制Wnt信号。该通路是毛细胞再生的潜在途径,并参与了神经母细胞、感觉上皮细胞的分化,耳基板的诱导和耳囊泡形态发生。
(5) BMP信号通路
BMPs(骨形态发生蛋白)是一个由20个成员组成的蛋白家族,是TGF β 超家族中最大的亚家族;调节了来自前感觉域支持细胞的命运图,并具有耳蜗中的非感觉细胞特性。它还参与了内耳的嵴和半规管的出现。BMP通路基因大多编码跨细胞膜运输所必需的蛋白,编码内耳毛细胞之间的大分子蛋白网络和在耳蜗外毛细胞中表达的蛋白。该通路还维持了毛细胞中的钙通道,刺激神经节神经元的突触传递。
(6) Shh信号通路
内耳腹侧极性与腹侧耳囊发育是通过Shh信号通路调控的。Shh信号通路也负责内耳的神经形成和耳囊腹侧生长。缺乏Shh信号通路会导致耳囊背腔极性改变以及球囊和耳蜗导管形成失败。GREB1L基因在Shh通路中发挥重要作用之一,能够在内耳早期发育过程中刺激VIII脑神经的发育。
(7) 凋亡通路
人体通过精准协调细胞增殖和细胞死亡来维持内耳正常的形态、发育和模式。在耳蜗管生长过程中,细胞死亡点保持保守(位于腹内侧位置)。在缺失相关信号的情况下,膜迷路会发生畸形。一些相关基因是线粒体DNA的一部分。12SrRNA和t-RNA基因具有通过活性氧(ROS)的产生调节线粒体的功能,从而介导细胞凋亡过程的功能。过量的ROS会导致高氧化应激,导致细胞死亡。在内耳形成过程中,过量的氧化应激可通过诱导耳蜗毛细胞凋亡而造成损伤。
四、NSHL的临床诊断及遗传检测
遗传性耳聋在临床和遗传学方面都存在异质性。研究策略的第一步是确定导致耳聋的临床原因。基因检测可以指导耳聋干预治疗,并为未来的研究提供见解(图3)。此外,借助目前具有成本效益的技术,可以面向大量受影响的家庭提供基因检测。
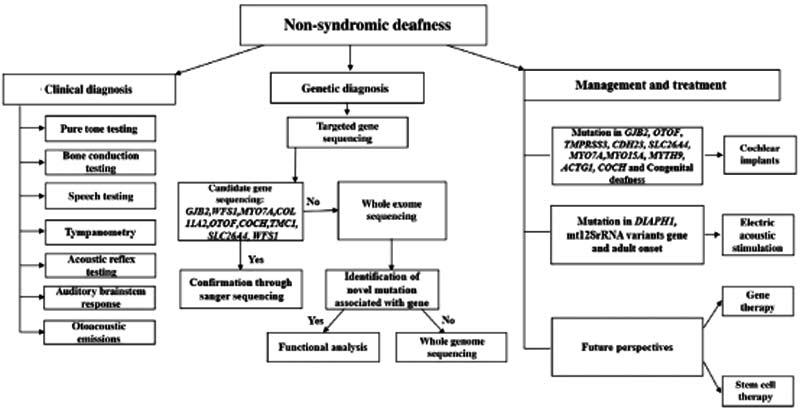
图3. NSHL相关临床诊断、基因检测以及治疗管理
尽管自20世纪90年代末以来,听力损失相关基因检测已经存在,但由于使用RFLP(限制性片段长度多态性)或Sanger测序的筛选通量较低,该检测仍然受到限制。为了克服RFLP的局限性,在pre-NGS时代,引入了更多的测序阵列和引物延伸微阵列,但这些技术仅限于检测已知的致病变异。
下一代测序(NGS)通过允许快速筛选整个外显子组或大基因面板,带来了对异质性疾病的分子诊断的变革。WES(全外显子组测序)和全基因组测序(WGS)似乎在未来几年中很有前途,但它有一定的局限性,如成本高、巨大数据分析的挑战和处理意义未知的变异。双亲性(当不止一个基因在同一个家族中分离时的一种情况)使得很难确定致病基因。相关致病性变异的类型和频率因不同的人群而异,一旦已知特定人群的常见致病变异,候选基因测序方法将为受影响的家庭提供帮助。尽管如此,Sanger测序仍然是确认这些变异的重要手段。
五、总结
本文总共描述了271篇论文中报道的2846例NSHL患者相关单基因致病性变异情况。共有98个基因的致病变异被认为是导致NSHL表型的单基因遗传致病原因。其中GJB2基因变异是最常见的NSHL遗传致病原因,在大量的NSHL患者中被报道。GJB2基因的致病变异能够导致Wnt信号通路受损,进而影响胚胎听觉系统的发育。综上所述,NSHL相关遗传致病原因的识别有助于疾病的管理和治疗。
参考文献
Sharma N, Kumari D, Panigrahi I, Khetarpal P. A systematic review of the monogenic causes of Non-Syndromic Hearing Loss (NSHL) and discussion of Current Diagnosis and Treatment options. Clin Genet. 2023 Jan;103(1):16-34. doi: 10.1111/cge.14228.
厦门助听器哪家好?厦门助听器-益耳助听器中心,真正近30年专业经验助听器验配,亲手验配5千名用户,世界六大品牌助听器,自有店面稳定经营厦门助听器专卖店,助您回归精彩有声世界。
相关阅读:耳聋基因检测
耳聋会不会遗传?
遗传性耳聋的遗传方式
如何看待儿童遗传性耳聋
我国最常见的耳聋基因-GJB2
GJB2耳聋基因听力损失为何差这多?























系列")